
In the search for stable and active catalysts, density
functional theory and machine learning-based models can
accelerate the screening of materials. While stability is conveniently
addressed on the bulk level of computation, the modeling of
catalytic activity requires expensive surface simulations. In this
work, we develop models for the surface adsorption energy of O
and OH intermediates across a consistent and extensive data set of
pure transition metal oxide surfaces. We show that adsorption
energies across metal oxidation states of +2 to +6 are well captured
from the metal−oxygen bond strength extracted from the bulk
level calculation. Specifically, we calculate the integrated crystal
orbital Hamiltonian population (ICOHP) of the metal−oxygen
bond in the bulk oxide and employ a simple normalization scheme
to obtain a strong correlation with the adsorption energetics. By combining our ICOHP descriptor with non-DFT features in a Gaussian Process regression (GPR) model, we achieve a high model accuracy with mean absolute errors of 0.166 and 0.219 eV for OH and O adsorption, respectively. By targeting the adsorption energy difference of the OH−OH adsorption with our GPR model, we predict the oxygen evolution reaction activity from bulk descriptors only. Furthermore, we utilize the strong correlation between the COHP and metal−oxygen bond lengths to rapidly predict the adsorption energetics and catalytic activity from the optimized bulk geometry. Our approach can enable an efficient search for active catalysts by eliminating the need for surface calculations in the initial screening phase. The full DFT dataset is available at http://www.catalysis-hub.org/publications/ComerGeneralized2024. 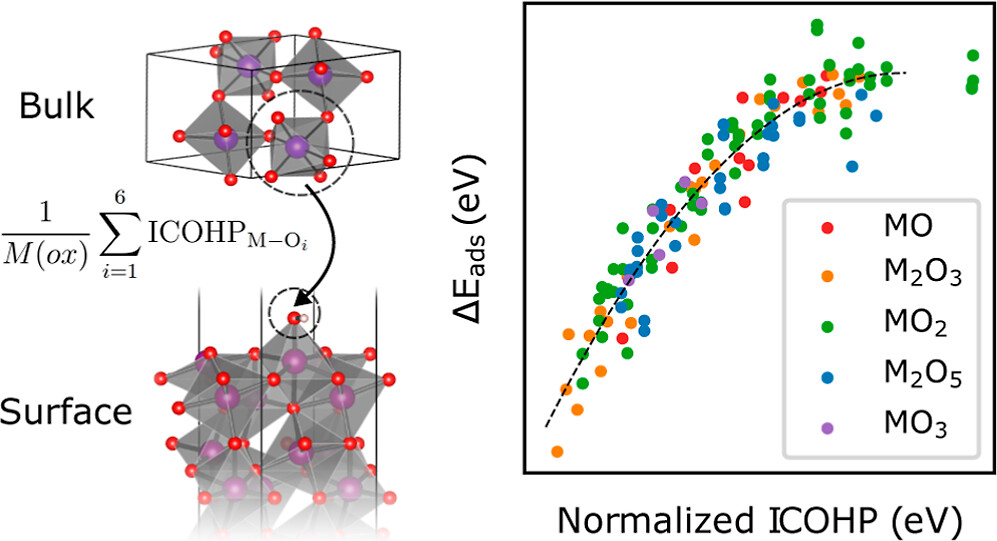
Prediction of O and OH Adsorption on Transition Metal Oxide Surfaces from Bulk Descriptors
Year of publication:
2024
Journal:
ACS Catalysis
Research Areas:
Funding sources: