
Despite developments in the field of transition metal-mediated catalysis, oxides, nitrides, carbides and sulfides still lack substantial understanding. Design strategies for these materials, which are often used in industrial processes, are now under development at SLAC.
In a recently published surface chemistry special feature in the Proceedings of the National Academy of Sciences, SLAC researchers at the Center for Sustainable Energy through Catalysis (SUNCAT) describe a blueprint for computer-aided design of catalytic materials.
The method involves the identification of catalytically relevant reaction steps on the active surfaces. The key reaction steps are then taken to the next level through identifying important trends for classes of catalysts across the periodic table.
Importantly, these trends can usually be represented by one or more simple descriptors. Identifying these descriptors is the main challenge. The publication takes an important step towards explaining how these descriptors can be established for large classes of reactions on transition metal surfaces. A typical descriptor might consist of the adsorption energies of the constituent atoms; adsorption energies of other intermediates and transition state energies are shown to scale with these. Establishing a set of simple descriptors allows for a fast computational screening for new and better catalyst leads.
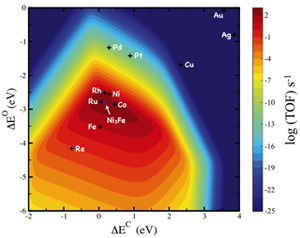
One example of this design strategy is the identification of two independent descriptors for the methanation reaction (hydrogenation of CO to methane to produce synthetic natural gas, for instance). It enabled the identification of nickel-iron catalysts as a cheaper and better alternative to pure nickel catalysts—a catalyst commonly used in industry (see Figure 1).