
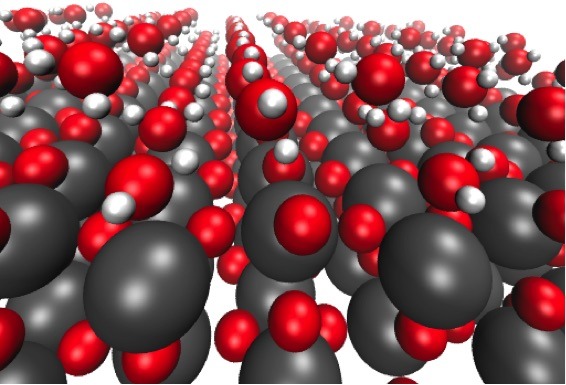
Formation of different water chains on defect free TiO2(110).
The presence of water is highly important in a large number of chemical and biological reactions. In many cases the reaction itself relies on the presence of water. Water can also act as an electrolyte in a large number of electrocatalytic reactions such as electrolysis and photolysis. For electrocatalytic and photocatalytic reactions taking place in water it is essential to include the effect of solvation of reactants, intermediates, and products at the surface. However, modeling water in computational studies is complex and requires very efficient functionals describing hydrogen bonding interactions, therefore it has only been done for a limited number of surfaces. For example, water splitting and the two sub-reactions, the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER), are in most cases studied theoretically without explicitly including the water.
We study the interface between adsorbed water and stoichiometric, defect-free (110) rutile oxide surfaces of TiO2, RuO2, and IrO2 in order to understand how water influences the stabilities of the intermediates of the oxygen evolution reaction (OER). In our model the water is treated as explicitly adsorbed H2O molecules, which are found to form two-dimensional water chains (layers) on all investigated oxide surfaces. The first chain formed by the most strongly bound H2O molecules is adsorbed on the five-fold coordinated surface metal atoms. The second chain is composed of less strongly bound H2O molecules binding to bridging oxygen atoms. The third chain interacts weakly and predominantly with the H2O molecules of the second layer, resembling bulk water. We find that the stability of the water layer close to the oxide surface is almost the same as the one found on flat metal surfaces, such as the Pt(111) surface, despite the highly different adsorption pattern of the water molecules. We show that the presence of a water network has some effect on the interaction of individual intermediates of the OER with the oxide surface. However, the theoretical OER overpotential remains almost unchanged in the case of RuO2 and IrO2, while it is increased by ~0.4 eV for TiO2.