
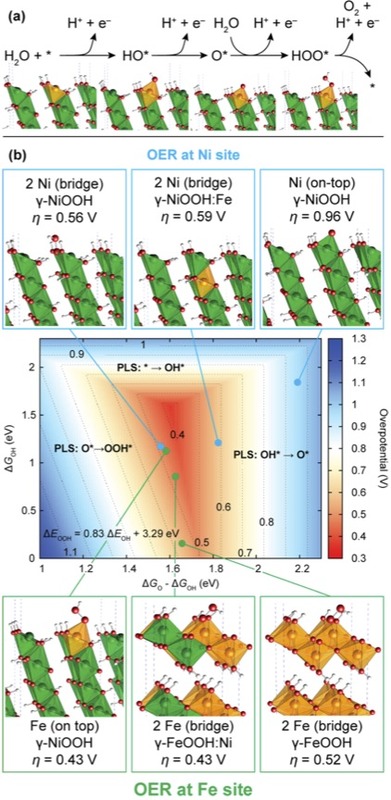
Theoretical OER overpotentials at Ni and Fe surface sites in pure and doped γ-NiOOH and γ-FeOOH model structures. (a) Proposed OER pathway with intermediates HO*, O* and HOO*, illustrated using the example of the on-top site at a substituted Fe surface atom in γ-NiOOH(0112̅).The binding energies of these species are used to estimate the OER overpotential. (b) OER activity volcano showing the overpotential as a function of Gibbs free energies of the reaction intermediates. Computed overpotentials are shown for the OER at Ni−Ni bridge and Fe on-top sites located in pure γ- NiOOH(011̅2) and in γ-NiOOH(011̅2) with Fe surface and subsurface doping, at a Ni on-top site in pure γ-NiOOH(0112̅), and at Fe−Fe bridge sites in pure and Ni-doped γ-FeOOH(010) (25% Ni in bulk unit cell). All corresponding model structures are shown with the intermediate whose formation is the potential limiting step (PLS) (HOO* in all cases except for the on-top Ni site in γ-NiOOH(0112̅) and γ-NiOOH(0112̅) with subsurface Fe, where formation of O* determines the overpotential).
The conversion of solar energy and water to hydrogen is a key element to a renewable fuel infrastructure, in which such 'renewable hydrogen' can be utilized in fuel cells for electric vehicles, or as reducing agent for the upgrading of biomass to hydrocarbon fuels or, ultimately, for the direct reduction of CO2 to fuels. The bottleneck for the efficiency of electrochemical and photoelectrochemical water splitting is the high overpotential required for the anodic half-reaction, the oxygen evolution reaction (OER). In contrast to many other catalytic reactions, e.g. in fuel cells, where thorough fundamental understanding of the interactions at the atomic scale that determine catalytic activity and catalyst stability in a corrosive environment has been achieved, such understanding, which could lead to breakthrough discoveries of new materials with significantly enhanced catalytic performance, is still lacking for the OER.
Recently, a team of researchers at the Joint Center for Artificial Photosynthesis (JCAP), SUNCAT, and the Stanford Synchrotron Radiation Lightsource (SSRL) investigated mixed Fe/Ni oxyhydroxide films, which are among the most active known catalysts based on earth-abundant elements for the OER in alkaline electrolytes. While it was known that the mixed Fe/Ni compounds exhibit up to 3 orders of magnitude higher OER activity than the pure Fe and Ni parent compounds, the origin of this enhancement was subject to speculation. The new study, which was published in Journal of the American Chemical Society, reveals the structure of Fe/Ni catalysts in their operating state during OER, and provides evidence for the first time on the nature of the active site, which is a Fe surface atom surrounded by neighboring Ni sites. This finding is in contrast to widely held views, according to which Ni, not Fe had been proposed to constitute the active sites in the mixed material.
Operando X-ray absorption spectroscopy (XAS) using the advanced X-ray spectrometer at SSRL beam line 6-2 indicated that for Fe contents up to ~25%, all Fe atoms substitute for Ni sites to form, under OER conditions, a Fe3+-containing Ni3+,4+ oxyhydroxide with unusually short Fe–O bond lengths. Using this knowledge of the structure of the operating catalyst, theoretical modeling of OER overpotentials was performed to gain insight into the origin of catalytic enhancement. DFT+U calculations, in which both Ni and Fe were tested as catalytic sites, reveal as a general trend that adsorbed OER intermediates become less stable at both sites in mixed materials compared to pure NiOOH and FeOOH. The energetics of the intermediates are thereby shifted further away from the optimum condition at Ni sites, while near-optimal energetics are achieved at Fe sites that have Ni neighbors.